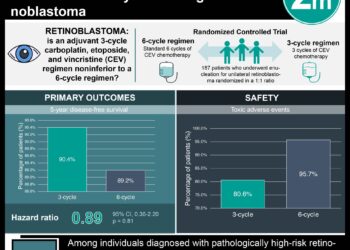
Patient Basics: Retinoblastoma
Originally published by Harvard Health.
What Is It?
Retinoblastoma is a form of cancer that develops on the retina. The retina is the structure at the back of the eye that senses light. It sends images to the brain which interprets them. In short, the retina allows us to see.
Although rare, retinoblastoma is the most common eye tumor in children. In most cases, it affects youngsters before age 5. It causes 5% of childhood blindness. But with treatment, the vast majority of patients maintain their sight.
About 40% of retinoblastoma cases are hereditary. This form of the disease usually affects children under age 2. It can affect one eye (unilateral) or both (bilateral).
All cases of bilateral retinoblastoma are hereditary. These cases can be associated with a tumor in the brain’s pineal gland. Unilateral retinoblastoma is usually not hereditary. It generally occurs in older children.
Children with retinoblastoma are more likely to develop other types of cancer later in life. The risk is higher in children with the hereditary type. Children treated with radiation therapy or certain types of chemotherapy also have a higher risk. Children who develop retinoblastoma in one eye have an increased risk of developing it in the other eye. They need frequent eye exams—even after treatment.
Doctors recommend that children with retinoblastoma get checked regularly for other cancers throughout their lives. Many of the second cancers that develop in long-term survivors of childhood retinoblastoma are caused by the radiation therapy used to treat the original cancer.
A specific gene leads to the development of retinoblastoma. In the hereditary form of the disease, all of the patient’s cells have a mutation, or change, in this gene. On its own, this single mutation doesn’t cause the disease. But if the patient develops a second mutation in a retina cell, the cancer can develop. If it does, both eyes are usually affected.
In the nonhereditary, or sporadic, form, both mutations occur by chance. It usually affects one eye.
The hereditary form of the disease—and the gene that causes it—can be associated with other types of cancer. These include cancers of the soft tissues or bone and an aggressive form of skin cancer.
Symptoms
The most common sign of retinoblastoma is a whitish-looking pupil. However, it does not always mean the child has the disease. Children with retinoblastoma also may have a crossed eye that turns out toward the ear or in toward the nose. But again, this is a common condition, one that’s likely to be noncancerous (benign).
Less common symptoms of retinoblastoma include
- redness and eye irritation that doesn’t go away
- differences in iris color and pupil size
- tearing
- cataracts
- bulging of the eyes.
Diagnosis
Newborns with a family history of retinoblastoma should be checked by an ophthalmologist (eye specialist) before leaving the hospital. But in most cases, a doctor diagnoses the condition after parents notice an abnormality and have the child’s eyes examined.
An ophthalmologist diagnoses retinoblastoma by doing a dilated-pupil examination. This involves viewing the retina with an indirect ophthalmoscope to see if a tumor exists. The indirect ophthalmoscope is different than the hand-held direct ophthalmoscope most doctors use to look inside the eye. It has more magnifying lenses and gives the ophthalmologist a clearer view of the entire retina.
This exam is usually done under general anesthesia. That way, the doctor can look carefully at the child’s retina. Sketches or photographs of the retina help “map” the tumor’s location.
Ultrasound, which uses sound waves to create images, often is done to measure larger tumors that make it difficult to see inside the eye. Next, either computed tomography (CT) scans or magnetic resonance imaging (MRI) scans may be done. By looking at the images they generate, doctors can determine whether the cancer has spread outside of the eye, into the brain, or to other parts of the body. If cancer has spread, additional tests may be needed.
Expected Duration
Retinoblastoma will continue to grow until it is treated.
Prevention
There is no known way to prevent retinoblastoma.
Because retinoblastoma may be hereditary, genetic testing is critical. Patients who carry the gene for the disease have an 80% chance of developing it. They also have a 50% chance of passing on the gene to a child. Siblings and children of retinoblastoma patients should be examined every two to four months during the first years of life.
Treatment
Treatment for retinoblastoma will depend on
- whether the disease affects one or both eyes
- the extent of the disease in the eye(s)
- whether vision can be saved
- whether the cancer has spread beyond the eye.
If the tumor is large, in one eye, and vision cannot be saved, the eye may be removed. This is a simple operation. About three to six weeks later, the child usually can be fitted with an artificial eye.
When tumors occur in one or both eyes and vision might be saved in one or both eyes, more conservative treatments may be considered. Radiation or chemotherapy may be used to shrink the tumors. Local treatments may then be used to eliminate the tumor and preserve vision. These may include brachytherapy, photocoagulation, and cryotherapy.
- Radiation can be an effective treatment for some patients. Retinoblastoma is very sensitive to radiation. However, it can damage the retina or other tissues in the eye. Radiation can also affect the growth of bone and other tissues near the eye. It may increase the risk of developing other cancers, too.
Two types of radiation may be used. External beam radiation involves focusing beams of radiation on the cancer from a source outside the body. Brachytherapy involves putting radioactive material into or near the tumor. In selected centers, proton therapy may be considered. It has the advantage of providing even more focused radiation therapy.
- Photocoagulation uses lasers to destroy the tumor.
- Cryotherapy uses extreme cold to freeze and destroy cancer cells. Doctors may choose cryotherapy for small tumors. To be effective, it usually has to be done several times. It is not used if the patient has several tumors.
- Chemotherapy involves receiving one or more anticancer drugs through an injection into a blood vessel. Retinoblastoma tends to resist chemotherapy, but it may be effective when combined with other treatments. For example, it may be used to shrink tumors to increase the chances of success with photocoagulation, cryotherapy, or brachytherapy. It is used commonly to treat a child whose tumor has spread beyond the eye. Chemotherapy also may be given to a child when the cancer has not spread beyond the eye, but when it has grown extensively within the eye, making it more likely to spread.
Retinoblastoma is a rare disease that requires specialized care. Seek treatment for your child at a center with staff experienced in treating it.
When To Call a Professional
If you see any abnormalities in your child’s eyes, take him or her to the doctor right away. You may be referred to a doctor who specializes in childhood eye diseases.
Prognosis
Early diagnosis and treatment are crucial to saving vision—and life. The outlook depends on how much the cancer has grown in and beyond the eye.
Nearly all children treated for retinoblastoma live at least five years. Children who are cancer-free after five years are generally considered cured. However, if left untreated, retinoblastoma is almost always fatal.
Survivors have an increased risk of developing a second, unrelated cancer. With regular follow-up, it may be caught and treated early.
Additional Information
American Cancer Society (ACS)
1599 Clifton Road, NE
Atlanta, GA 30329-4251
Toll-Free: 800-227-2345
http://www.cancer.org/
National Cancer Institute (NCI) and the National Eye Institute
U.S. National Institutes of Health
Public Inquiries Office
Building 31, Room 10A03
31 Center Drive, MSC 8322
Bethesda, MD 20892-2580
Phone: 301-435-3848
Toll-Free: 800-422-6237
TTY: 800-332-8615
http://www.nci.nih.gov/ (National Cancer Institute)
http://www.nei.nih.gov/ (National Eye Institute)
RelatedReports